Selection of features

Command based input
MolTwister runs in a command line shell, either Linux or MacOS. Commands are first interpreted by the MolTwister command processor. If the command is not found, it is sent to regular command line shell. Hence, you have all the standard shell commands available within MolTwister, such as ls, cd, mkdir. Moreover, other software like vim can also be executed from within MolTwister. MolTwister commands are primarily used to build molecular systems, as well as to perform post processing on data from molecular dynamics simulations.

3D Visualization of model
After having started MolTwister, a 3D visualization window is shown. This will show a 3D representation of the currently constructed molecular systems model. Atoms are added, moved and configured using the command line shell.
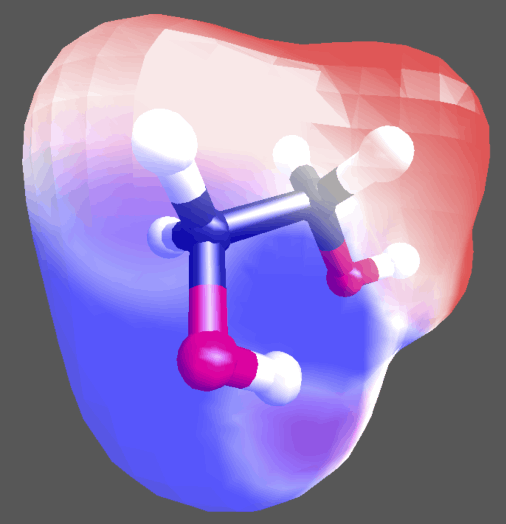
Visualize charge distributions
For a limited amount of atoms, charges can be assigned and an iso-surface representation of the charge distribution can be visualized.
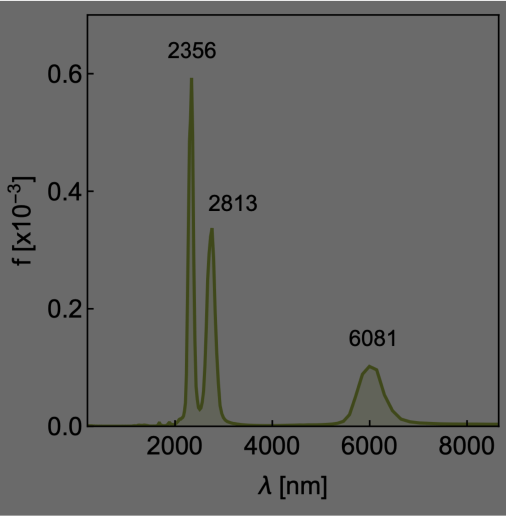
Analyze MD results
Several analysis can be performed on molecular data and trajectories from MD simulations. For example, it is possible to
* Calculate number, mass and charge density profiles
* Calculate dihedral distributions
* Calculate dipole moments and dipole moment profiles
* Calculate hydrogen bond statistics
* Calculate the mean square displacement
* Calculate pair correlation functions
* Calculate velocity auto correlation functions
* Calculate the vibrational (phonon) density of states
* Perform simple charge balancing of surface slabs
Import & Output
It is possible to read XYZ files (also containing several frames), PDB files (currently only supports loading of atomic coordinates) and DCD files into the program. The program can export to XYZ files and PDB files. It can also perform modifications of DCD files.
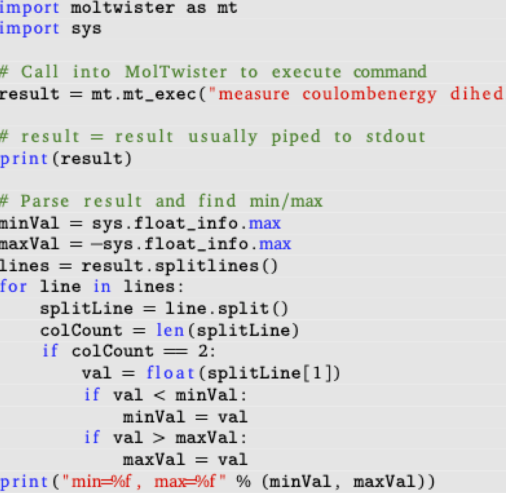
Scripting
There are two options for scripting.
* Write script files containing sequences of shell commands to execute
* Write python notebooks that can format and execute shell commands and similar